
What is Good Pharmacovigilance Practice (GVP)?
Good Pharmacovigilance Practice (GVP) is a set of measures drawn up to facilitate the performance of pharmacovigilance in the European Union (EU). It represents the minimum standard for monitoring the safety of medicines available to the public. These practices consist of specific guidelines that help track and report adverse drug reactions (ADRs) and other safety concerns related to pharmaceutical products.
GVP applies to marketing authorization holders, the European Medicines Agency (EMA), and medicines regulatory authorities in EU Member States. These guidelines cover medicines authorized centrally via the Agency as well as those authorized at national level, establishing consistent safety standards that protect patients worldwide.
The primary purpose of GVP is to prevent adverse effects from the authorized use of pharmaceutical products and to promote their safe and effective use. According to the World Health Organization (WHO), any newly developed drug must meet three critical criteria prior to approval – good quality, effectiveness, and safety for intended purposes.
Key aspects covered under GVP include monitoring interactions of medicines, abuse and misuse of medicines, counterfeit medicines, medication errors, adverse drug reactions and events, and lack of efficacy. The scope extends beyond traditional pharmaceuticals to include vaccines, blood products, herbals, medical devices, biological, and traditional/complementary substances.
In Canada, establishments selling drug products must comply with the Food and Drugs Act and Food and Drug Regulations, which require drug establishments to monitor the ongoing safety of their products and report any adverse drug reactions to the federal government. These establishments must maintain ADR records and annual summary reports for 25 years, allowing authorities to monitor drug reactions over time and request label changes when necessary.
Similarly, the FDA provides guidance on good pharmacovigilance practices, specifically focusing on safety signal identification, pharmacoepidemiologic assessment and safety signal interpretation, and pharmacovigilance plan development. Furthermore, the EMA and FDA participate in collaborative processes for information-sharing on GVP through a ‘cluster’ relationship formed in 2014.
Even though approved pharmaceuticals undergo rigorous testing before market authorization, most tests are conducted with relatively small numbers of subjects under controlled circumstances. Through good pharmacovigilance practices, pharmaceutical companies can identify issues missed during initial testing and deliver critical safety information to healthcare professionals and the public.
Regulatory authorities in different countries conduct inspections to determine whether companies comply with pharmacovigilance obligations. For instance, the UK’s MHRA has been conducting statutory GPvP inspections since 2003, while Canada implements a GVP inspection program to ensure establishments continually evaluate drug safety and effectiveness.
Why is GVP important in pharmacovigilance?
GVP plays a critical role in protecting patients and public health by establishing standardized frameworks for monitoring medicine safety. The primary importance of GVP lies in its ability to identify and evaluate potential risks associated with medications, thereby protecting patients from adverse reactions. Through early identification and rapid response to safety concerns, GVP minimizes the impact on public health.
One of the fundamental reasons GVP is essential involves its contribution to healthcare decision-making. By providing healthcare professionals with comprehensive safety information, GVP enables informed decisions about prescribing and administering medications. Moreover, GVP data identifies areas for further research and development, ultimately leading to safer and more effective medications.
Clinical trials can only detect adverse effects in relatively small populations under controlled conditions. As noted by WHO, these trials involve studying products in a limited number of selected individuals for short periods. Consequently, certain side effects may only emerge once medications have been used by diverse populations, including people with concurrent diseases, over extended periods.
The importance of GVP extends to the following key areas:
- Risk Management: GVP mandates detailed Risk Management Plans for medicinal products, outlining known risks and proposing preventive measures
- Signal Detection: Continuous surveillance of data sources enables identification of new safety signals before they become significant issues
- Benefit-Risk Assessment: Ongoing evaluation ensures emerging risks are properly weighed against benefits throughout a product’s lifecycle
- Transparent Communication: GVP emphasizes clear communication of drug risks to healthcare professionals, patients, and the public
Additionally, GVP provides a structured approach for collecting comprehensive data from various sources, including healthcare professionals, patients, and scientific literature. This systematic data gathering facilitates efficient retrieval and analysis through centralized databases.
GVP requirements have evolved alongside increased regulatory oversight, highlighting their significance in both premarket review and post-market surveillance. This expansion in scope, complexity, and responsibility has made high-quality pharmacovigilance critical to successful pharmaceutical development and monitoring.
The thalidomide tragedy of the past served as a catalyst for strengthening regulatory frameworks on drug safety. From this incident, spontaneous reporting of adverse drug reactions became systematic, organized, and regulated, underscoring the necessity of robust pharmacovigilance practices to prevent similar public health crises.
How does GVP work in drug safety monitoring?
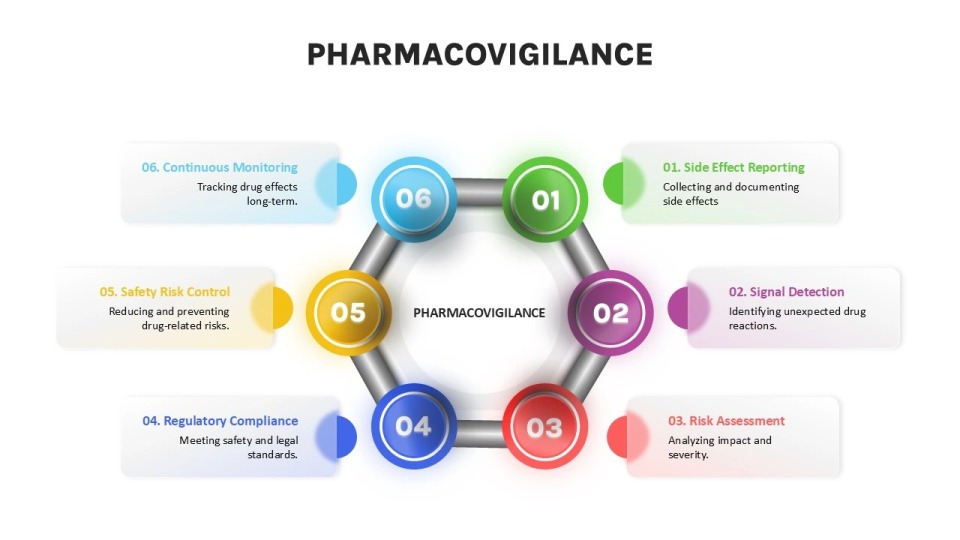
Image Source: SlideBazaar
GVP implementation in drug safety monitoring follows a structured approach consisting of several interconnected processes. This systematic methodology enables regulatory authorities and marketing authorization holders to identify, evaluate, and mitigate risks associated with medicinal products throughout their lifecycle.
Signal detection and validation
Signal detection forms the foundation of the GVP safety monitoring system. A signal is defined as information suggesting a new potentially causal association, or a new aspect of a known association, between an intervention and an event that requires verification. The process involves continuous monitoring of data from multiple sources, including spontaneous reports, clinical trials, scientific literature, and active surveillance systems.
Once identified, signals undergo validation to confirm whether available documentation contains sufficient evidence demonstrating a new potentially causal association. Validation considers three primary elements: previous awareness of the issue, strength of evidence, and clinical relevance. This process determines if further assessment is warranted based on scientific plausibility and reproducibility across data sources.
Risk assessment and minimization
After validation, signals undergo assessment to evaluate their impact on the product’s benefit-risk profile. This involves examining additional information sources to establish or refute causal relationships. Signals suggesting risks with significant patient or public health impact receive prioritization for urgent attention and management.
Risk minimization measures (RMMs) are subsequently implemented to prevent or reduce adverse reactions. These fall into two categories:
- Routine measures: Applied to all medicinal products, including the Summary of Product Characteristics (SmPC), package leaflets, labeling, pack size, and legal status
- Additional measures: Implemented when routine measures are insufficient, such as educational materials, controlled access programs, or pregnancy prevention protocols
The effectiveness of these measures must be evaluated throughout the product lifecycle to determine if adaptations are necessary.
Reporting and documentation
GVP mandates comprehensive documentation of all pharmacovigilance activities. Marketing authorization holders must submit Periodic Safety Update Reports (PSURs) providing comprehensive reviews of product safety profiles, including newly identified risks and actions taken. Additionally, emerging safety issues require immediate written notification to competent authorities.
Regulatory bodies expect a fully traceable signal management process documented at every stage. This documentation serves as evidence of compliance with GVP requirements and allows authorities to evaluate the effectiveness of safety monitoring activities.
Key components of GVP guidelines
GVP guidelines incorporate several standardized documents that form the backbone of effective pharmacovigilance systems. These components ensure consistent monitoring, assessment, and communication of medicinal product safety throughout the product lifecycle.
Risk Management Plan (RMP)
The Risk Management Plan documents the risk management system necessary to identify, characterize, and minimize a medicinal product’s important risks. An RMP contains three core elements: safety specification focusing on identified and potential risks, pharmacovigilance plan outlining activities to further characterize these risks, and risk minimization plan describing measures to mitigate risks. As a dynamic document, the RMP evolves throughout the product’s lifecycle as knowledge about its safety profile increases. Companies must submit an RMP when applying for marketing authorization.
Periodic Safety Update Report (PSUR)
PSURs provide comprehensive evaluations of the risk-benefit balance of medicinal products at defined post-authorization intervals. These documents present concise, critical analyzes of new safety information in the context of cumulative data. PSURs incorporate data from various sources, including clinical trials, spontaneous reports, active surveillance systems, and scientific literature. Their format follows the structure described in Commission Implementing Regulation (EU) No 520/2012.
Development Safety Update Report (DSUR)
The DSUR serves as a common standard for periodic reporting on drugs under development across ICH regions. Submitted annually, it replaces the US IND Annual Report and EU Annual Safety Report. The DSUR provides thoughtful review of pertinent safety information collected during the reporting period, examining whether new data aligns with previous knowledge.
Company Core Safety Information (CCSI)
CCSI represents the minimum essential safety information that manufacturers require to be listed in all countries where their drug is marketed. Contained within the Company Core Data Sheet, CCSI excludes extraneous or inadequately substantiated information. It serves as reference for determining “listed” and “unlisted” adverse reactions for periodic reporting purposes.
Summary of Product Characteristics (SmPC)
The SmPC provides essential information about a medicinal product, including indications, dosage, contraindications, and potential side effects. It functions as a key reference document for healthcare professionals and patients, forming part of routine risk minimization measures.
How GVP fits into the global pharmacovigilance system
The global pharmacovigilance landscape relies on harmonized regulatory frameworks, with Good Pharmacovigilance Practice (GVP) serving as a foundational standard that transcends geographical boundaries. Originally established by the European Medicines Agency (EMA) in 2012, GVP has influenced pharmacovigilance practices worldwide.
Globally, three major regulatory authorities drive pharmacovigilance regulations: the EMA, US Food and Drug Administration (FDA), and Japan’s Pharmaceuticals and Medical Devices Agency (PMDA). These agencies collaborate through various international initiatives to ensure consistent medicine safety standards. Notably, the FDA and EMA formed a ‘cluster’ relationship in 2014 specifically for information-sharing on GVP.
FDA engages with stakeholders internationally through organizations including the International Council for Harmonization (ICH), International Pharmaceutical Regulators Program (IPRP), and International Coalition of Medicines Regulatory Authorities (ICMRA). This collaboration advances pharmaceutical safety and effectiveness across borders.
Many nations have adapted GVP standards to their regulatory frameworks. Australia follows Europe’s Black Triangle scheme, Saudi Arabia has adopted EU GVP, and Chile tracks EMA practices particularly during the COVID pandemic.
The World Health Organization established its Program for International Drug Monitoring in 1968, with the Uppsala Monitoring Center maintaining VigiBase, the global database of adverse event reports. This infrastructure enables worldwide sharing of safety signals, demonstrating how GVP principles operate within an interconnected global pharmacovigilance network.
FAQs
Q1. What is the purpose of Good Pharmacovigilance Practice (GVP)? GVP aims to prevent adverse effects from authorized pharmaceutical products and promote their safe and effective use. It establishes a standardized framework for monitoring medicine safety, identifying potential risks, and protecting public health.
Q2. How does GVP contribute to drug safety monitoring? GVP works through a structured approach involving signal detection, risk assessment, and reporting. It includes continuous monitoring of data from various sources, evaluating potential risks, implementing risk minimization measures, and documenting all pharmacovigilance activities.
Q3. What are the key components of GVP guidelines? The main components include the Risk Management Plan (RMP), Periodic Safety Update Report (PSUR), Development Safety Update Report (DSUR), Company Core Safety Information (CCSI), and Summary of Product Characteristics (SmPC). These documents ensure consistent monitoring and communication of medicinal product safety.
Q4. How does GVP fit into the global pharmacovigilance system? GVP serves as a foundational standard in the global pharmacovigilance landscape, influencing practices worldwide. Major regulatory authorities like EMA, FDA, and PMDA collaborate through international initiatives to ensure consistent medicine safety standards across borders.
Q5. Who is required to comply with GVP? GVP applies to marketing authorization holders, the European Medicines Agency (EMA), and medicines regulatory authorities in EU Member States. It covers medicines authorized centrally via the Agency as well as those authorized at the national level.